


Our Research
Overview
Ryvu Therapeutics discovers and develops small molecule therapies that address high value emerging targets in oncology.
Our pipeline is built from internally-discovered candidates which make use of diverse therapeutic mechanisms, including programs directed at targets in the areas of transcriptional regulation, synthetic lethality and immuno-oncology.
Ryvu’s most advanced programs include RVU120, a selective CDK8/CDK19 kinase inhibitor with potential to treat hematological malignancies and solid tumors, currently in Phase II development for the treatment of patients with relapsed/refractory acute myeloid leukemia (r/r AML) in combination with venetoclax.
SEL24 (MEN1703) is a dual PIM/FLT3 kinase inhibitor in clinical development for the treatment of relapsed/refractory diffuse large B-cell lymphoma (DLBCL).. Additional programs include synthetic lethality candidates directed at WRN and MTAP deletions as well as immuno-oncology programs focused on STING agonists and HPK1.
- Clinical Projects
- Immuno-oncology Collaborations
- Synthetic Lethality
Clinical Projects
-
RVU120
CDK8 is a kinase submodule of the mediator complex, involved in both transcriptional activation and repression. CDK8- mediator complex integrates basal transcriptional machinery with the activity of oncogenic transcriptional and epigenetic factors. Targeting CDK8 and...
-
SEL24 (MEN1703)
SEL24 (MEN1703) is a selective, dual inhibitor of PIM and FLT3 kinases, two enzymes that are strongly implicated in malignant transformation of hematopoietic cells. The compound is a novel small molecule discovered by Ryvu, and...


Synthetic Lethality
One of the challenges in the development of new oncology therapeutics is the limited number of protein targets known to be addressable with small molecules. Synthetic lethality allows the identification of novel protein targets and mechanisms of cancer cell sensitivity that can be used therapeutically.
Compounds active on specific synthetic lethal targets selectively kill tumor cells of a precisely defined model system, characterized by a specific genetic, epigenetic or metabolic characteristic. This is the approach actively investigated at Ryvu that allows the generation of compounds with highly specific and well-characterized activity.

-
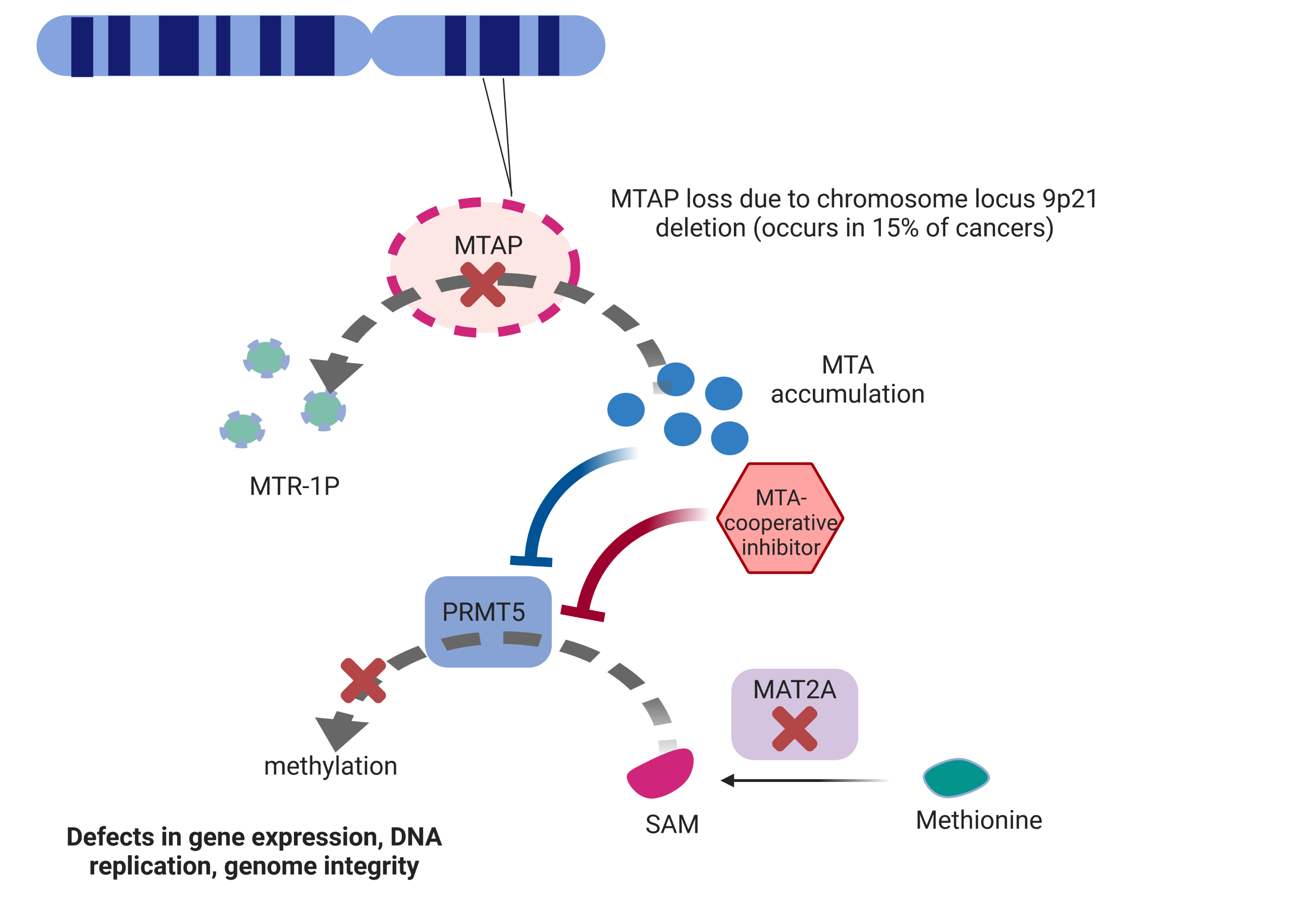
MTA-COOPERATIVE PRMT5 INHIBITOR
Deletion of 9p21 locus is one of the most frequent genetic alterations associated with human cancers. Apart from tumour suppressor CDKN2A, locus 9p21 contains the gene coding methylthioadenosine phosphorylase (MTAP), the only enzyme capable of...
-
WRN INHIBITOR
Werner Syndrome helicase (WRN) is a member of the RecQ helicase family and plays an important role in controlling DNA repair mechanisms and maintaining integrity of the genome. WRN helicase has been identified to be...
Novel Targets
Beyond publicly disclosed programs, Ryvu is working on candidates that address novel targets in the areas of synthetic lethality, immuno-oncology and immunometabolism to generate new therapeutics with first-in-class or best-in-class potential.
For the candidates, Ryvu is utilizing a proprietary drug discovery approach combining internally developed expertise and new sophisticated tools and technologies to identify small molecule compounds with favorable profiles and activity.
Immuno-Oncology and other R&D Collaborations
In November 2022, Ryvu Therapeutics and BioNTech have entered into a multi-target research collaboration for several small molecule immunotherapy programs. BioNTech and Ryvu will jointly undertake drug discovery and research projects to develop multiple small molecule programs directed at exclusive targets selected by BioNTech, primarily focused on immune modulation within oncology, with potential applications in other disease areas.
In 2013, Ryvu and Merck KGaA initiated a joint research strategy involving intensive work on multiple targets and several chemical series. The collaboration has successfully completed two milestones and has been expanded over time with additional targets.
The aim of the partnership with Merck is the development of new anticancer therapeutics acting on diverse biological targets associated with aberrant metabolic pathways in cancer cells. Dependence on specific metabolic pathways is a feature of various types of cancer, therefore, targeting this vulnerability has many potential applications in a broad range of patient populations.
The collaboration has resulted in scientific achievements, the joint filing of patent applications and data presentations at international scientific conferences.

In 2022 Ryvu and Exelixis signed an exclusive license agreement focused on the development of novel targeted therapies utilizing Ryvu’s STING (STimulator of INterferon Genes) technology. The agreement expands Exelixis’ portfolio of biotherapeutics by combining Ryvu’s proprietary small molecule STING agonists and STING biology know-how with Exelixis’ network of expertise and resources in antibody engineering, antibody-drug conjugate (ADC) technologies, and proven history of developing and commercializing oncology therapeutics.
Publications
European Hematology Association (EHA) Congress, June 13 -16, 2024
-
RVU120, a first-in-class CDK8 inhibitor for the treatment of relapsed/refractory AML and high-risk MDS: preliminary results from two ongoing studies.

-
Synergistic potential of RVU120, a first-in-class CDK8/CDK19 inhibitor, with venetoclax in AML: preclinical and initial clinical insights.
-
CDK8/19 Inhibition: A Promising Therapeutic Strategy in Myeloproliferative Neoplasms.
To download chosen material please fill in form below:
To download chosen material please fill in form below:
To download chosen material please fill in form below:
American Association for Cancer Research (AACR) Annual Meeting, April 5-10, 2024
-
Discovery of novel MTA-cooperative PRMT5 inhibitors as targeted therapeutics for MTAP-deleted cancers
-
Discovery of WRN inhibitors as targeted therapy in the treatment of microsatellite unstable (MSI-H) tumors
-
A Comprehensive Platform for Identification of KRAS-specific Synthetic Lethal Targets using Patient-Derived Cells
-
MEN1703/SEL24 exhibits promising antitumoral activity in preclinical models of myelofibrosis both as a single agent and combined with ruxolitinib.
-
Combination JAK1/2 and CDK8/19 inhibition demonstrates enhanced efficacy in myeloproliferative neoplasms.
To download chosen material please fill in form below:
To download chosen material please fill in form below:
To download chosen material please fill in form below:
To download chosen material please fill in form below:
To download chosen material please fill in form below:
American Society of Hematology (ASH) Annual Meeting, December 9 –12, 2023
-
Safety and Efficacy Results from CLI120-001 a Phase 1 Study in RR-AML and HRMDS: Update from Higher Dose Levels
-
Preclinical and Clinical Evidence for Erythroid-Stimulating Activity of RVU120 CDK8/19 Inhibitor in AML and MDS
-
Novel Clinically Useful Inhibitor of Mediator Complex, RVU120, Relieves Differentiation Block in MDS/AML
-
Targeting CDK8/CDK19 to Disrupt Leukemic Stem Cell-like Population in Acute Myeloid Leukemia: Exploring RVU120 As a Promising Frontline Therapy
-
Mediator Kinase/CDK8 Inhibition as a Strategy to Improve FLT3 Inhibitor Activity in Acute Myeloid Leukemia
To download chosen material please fill in form below:
To download chosen material please fill in form below:
To download chosen material please fill in form below:
To download chosen material please fill in form below:
To download chosen material please fill in form below:
San Antonio Breast Cancer Symposium (SABCS), December 5-9, 2023
-
Synergistic Activity of CDK8/19 Inhibitor RVU120 and MEK Inhibitors in Hormone-Negative Breast Cancer: Implications for Targeted Therapy